FiRE
This project is maintained by princethewinner
FiRE - Finder of Rare Entities
Update: FiRE is now available via cran. install FiRE using
install.packages('FiRE')
Contents
Introduction
External dependencies
Installation
Python Package
-Prerequisites
-Installation Steps
-Usage
R Package
-Prerequisites
-Installation Steps
-Usage
Publication
Copyright
Patent
Introduction
Tested on Ubuntu 14.04 and Ubuntu 16.04.
All results in manuscript have been generated using python module.
FiRE is available for python and R. Required versions and modules for both are as mentioned below. cpp modules are necessary for both of them.
External Dependencies
Following packages are required to run/install the FiRE software.
Required cpp modules
g++ >= 4.8.4
boost >= 1.54.0
FiRE only needs <boost/random.hpp> from boost. So, full installation is not necessary. It can be downloaded from boost.org and used as is.
Installation
[sudo] ./INSTALL [ --boost-path <boost-path> | --log-file <log-file> | --inplace | --py | --R | --help ]
[sudo] ./UNINSTALL_python
[sudo] ./UNINSTALL_R
--boost-path <boost-path> : python : Path to boost-library, if boost is not installed at default location, this value needs to be provided.
--inplace : python : Required only for python, if set, inplace build will be run and resulting lib will be stored in python/FiRE.
--log-file <log-file> : python : Required only for python, ignored with --inplace set.
--py : python : Install FiRE in python environment.
--R : R : Install FiRE in R environment.
--help : python | R : Display this help.
Info:
UNINSTALL_[python | R] files are generated upon installation.
Typically, FiRE module takes a few seconds to install. A snippet of installation time taken by FiRE (in seconds) on a machine with Intel® Core™ i5-7200U (CPU @ 2.50GHz × 4), with 8GB memory, and OS Ubuntu 16.04 LTS is as follows
real 2.92
user 2.73
sys 0.18
Python Package
Prerequisites
Required python modules
python 2.7
# [EDIT] python 3 can also be used with standard installation.
# however, with --inplace option uninstallation may fail.
# As a workround generated .so file can be removed manually if uninstallation of FiRE is desired.
For FiRE module
cython >= 0.23.4
distutils >= 2.7.12
For preprocessing
numpy >= 1.13.3
pandas >= 0.20.3
statsmodels >= 0.8.0
For demo
gzip >= 1.2.11 (zlib)
scipy >= 1.1.0
matplotlib >= 2.1.0
cmocean >= 1.2
sklearn >= 0.19.1
Installation Steps
with virtual environment avoid using sudo. (Thanks to chenxofhit)
[sudo] chomd +x ./INSTALL
If boost is installed at default location
[sudo] ./INSTALL --py
If boost is installed at custom location
[sudo] ./INSTALL --boost-path <full-path> --py
Example:
[sudo] ./INSTALL --boost-path $HOME/boost/boost_1_54_0 --py
Above installation steps will generate fireInstall.log file. It is advisable to keep this file, since it will be needed for uninstallation. Name of the log file can be modified during installation.
./INSTALL --log-file <log-file-name> --py
Above steps will install FiRE at the default location.
For inplace installation
./INSTALL --inplace --py
Uninstallation of FiRE Software.
[sudo] ./UNINSTALL_python
Usage
Run demo from FiRE directory as follows
python example/jurkat_simulation.py
Since data (data/jurkat_two_species_1580.txt.gz) is large, this may require large amount of RAM to load and pre-process. We have also providee pre-processed data (data/preprocessedData_jurkat_two_species_1580.txt.gz). Pre-processing was done using the script present in utils/preprocess.py. Demo using this data as follows
python example/jurkat_simulation_small.py
Small demo takes seconds to complete. Exact time taken by the demo on a machine with Intel® Core™ i5-7200U (CPU @ 2.50GHz × 4), with 8GB memory, and OS Ubuntu 16.04 LTS is as follows
Loading preprocessed Data : 1.850723s
Running FiRE : 1.134673s
Total Demo time:
real 4.33
user 3.55
sys 0.76
Step-by-step description of full demo (example/jurkat_simulation.py) is as follows
-
Load libraries
```python import sys sys.path.append(‘utils’)
import numpy as np import gzip from scipy import stats
import preprocess as pp import misc import FiRE
2. <h4>Load Data in current environment.</h4>
```python
#Data matrix should only consist of values where rows represent cells and columns represent genes.
with gzip.GzipFile('data/jurkat_two_species_1580.txt.gz', 'r') as fid:
data = np.genfromtxt(fid)
data = data.T #Samples * Features
labels = np.genfromtxt('data/labels_jurkat_two_species_1580.txt', dtype=np.int) #Cells with label '1' represent abundant, while cells with label '2' represent rare.
Data Pre-processing
-
Call function ranger_preprocess for selecting thousand variable genes.
```python
#Genes genes = np.arange(1, data.shape[1]+1) #It can be replaced with original gene names
#Filter top 1k genes preprocessedData, selGenes = pp.ranger_preprocess(data, genes, optionToSave=True, dataSave=outputFolder)
|Parameter | Description | Required or Optional| Datatype | Default Value |
| -----:| -----:| -----:|-----:|-----:|
|data | Data for processing | Required | `np.array [nCells, nGenes]` | - |
|genes | Names of Genes | Required | `np.array [nGenes]` | - |
|ngenes_keep | Number of genes to keep | Optional | `integer` | 1000 |
|dataSave | Path to save results | Optional | `string` | Current working Directory (Used only when optionToSave is True) |
|optionToSave | Save processed output or not | Optional | `boolean` | False(Does not save) |
|minLibSize | Minimum number of expressed features | Optional | `integer` | 0 |
|verbose | Display progress | Optional | `boolean` | True(Prints intermediate results) |
```python
'''
Returned Value :
preprocessedData : processed data matrix (log2 transformed) : np.array [nCells, nVariableGenes]
selGenes : Names of thousand variable genes selected : np.array [nVariableGenes]
'''
-
Create model of FiRE.
model = FiRE.FiRE(L=100, M=50, H=1017881, seed=5489, verbose=0)
| Parameter | Description | Required or Optional | Datatype | Default Value |
|---|---|---|---|---|
| L | Total number of estimators | Required | int |
- |
| M | Number of features to be randomly sampled for each estimator | Required | int |
- |
| H | Number of bins in hash table | Optional | int |
1017881 |
| seed | Seed for random number generator | Optional | unsigned int |
5489 |
| verbose | Controls verbosity of program at run time (0/1) | Optional | int |
0 (silent) |
-
Apply model to the above dataset.
model.fit(preprocessedData) -
Calculate FiRE score of every cell.
```python score = np.array(model.score(preprocessedData)) ‘’’ Returned Value : score : FiRE score of every cell : np.array[nCells]
Higher values of FiRE score represent rare cells. ‘’’
7. <h4>Select cells with higher values of FiRE score, that satisfy IQR-based thresholding criteria.</h4>
```python
q3 = np.percentile(score, 75)
iqr = stats.iqr(score)
th = q3 + 1.5*iqr
indIqr = np.where(score >= th)[0]
dataSel = preprocessedData[indIqr,:] #Select subset of rare cells
#Create a file with binary predictions
predictions = np.zeros(data.shape[0])
predictions[indIqr] = 1 #Replace predictions for rare cells with '1'.
-
Access to model parameters.
Sampled dimensions can be accessed via
# type : 2d list # shape : L x M model.dimsChosen thresholds can be accessed via
# type : 2d list # shape : L x M model.thresholds
Weights can be accessed via
# type : 2d list
# shape : L X M
model.weights
Hash tables can be accessed via
# type : 3d list
# shape : L x H x <dynamic>
# <dynamic> : as per number of samples in a bin (H) for a given estimator (L).
model.bins
-
FiRE recovers artifitially planted rare cells (Figure).
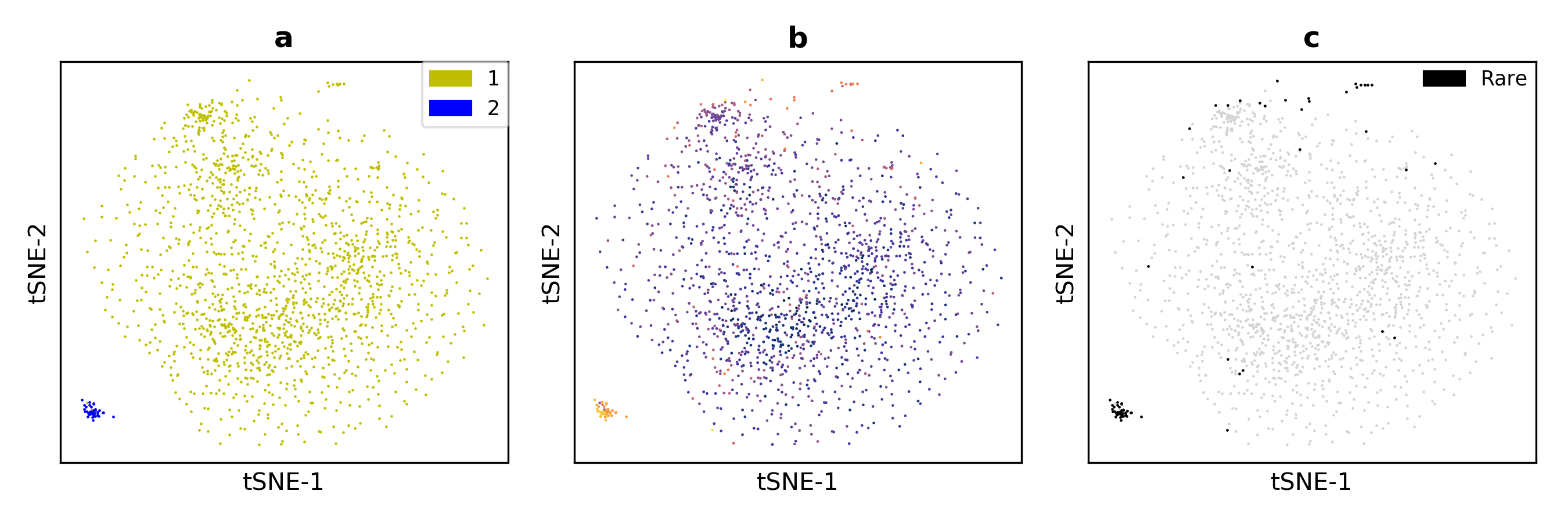
(a) t-SNE based 2D embedding of the cells with color-coded identities (b) FiRE score intensities plotted on the t-SNE based 2D map. (c) Rare cells detected by FiRE.
R Package
Prerequisites
Required R modules
R >= 3.2.0
For FiRE module
Rcpp >= 0.12.19
BH >= 1.66
For preprocessing and demo
Matrix >= 1.2.14
plyr >= 1.8.4
Installation Steps
[sudo] chomd +x ./INSTALL
Installation of FiRE Software.
[sudo] ./INSTALL --R
Uninstallation of FiRE Software.
[sudo] ./UNINSTALL_R
Usage
Run demo from FiRE directory as follows
Rscript example/jurkat_simulation.R
Since data (data/jurkat_two_species_1580.txt.gz) is large, this may require large amount of RAM to load and pre-process. We have also providee pre-processed data (data/preprocessedData_jurkat_two_species_1580.txt.gz). Pre-processing was done using the script present in utils/preprocess.R. Demo using this data as follows
Rscript example/jurkat_simulation_small.R
Small demo takes seconds to complete. Exact time taken by the demo on a machine with Intel® Core™ i5-7200U (CPU @ 2.50GHz × 4), with 8GB memory, and OS Ubuntu 16.04 LTS is as follows
Total Demo time:
real 4.11
user 3.16
sys 1.13
Step-by-step description of full demo (example/jurkat_simulation.R) is as follows
-
Load libraries
library('FiRE') source('utils/preprocess.R') -
Load Data in current environment.
```R #Read data data <- read.table(gzfile(‘data/jurkat_two_species_1580.txt.gz’)) data <- t(data) #Samples * Features
#Read Labels labels <- read.table(‘data/labels_jurkat_two_species_1580.txt’) #Cells with label ‘1’ represent abundant, while cells with label ‘2’ represent rare.
#Genes genes <- c(1:dim(data)[2]) #It can be replaced with original gene names
data_mat <- list(mat=data, gene_symbols=genes)
3. <h4>Call function ranger_preprocess for selecting thousand variable genes.</h4>
```R
preprocessedList <- ranger_preprocess(data_mat)
preprocessedData <- as.matrix(preprocessedList$preprocessedData)
| Parameter | Description | Required or Optional | Datatype | Default Value |
|---|---|---|---|---|
| data_mat | List consisting of data for processing and gene symbols | Required | list(mat=data, gene_symbols=genes) |
- |
| ngenes_keep | Number of genes to keep | Optional | integer |
1000 |
| dataSave | Path to save results | Optional | string |
Current working Directory (Used only when optionToSave is True) |
| optionToSave | Save processed output or not | Optional | boolean |
False(Does not save) |
| minLibSize | Minimum number of expressed features | Optional | integer |
0 |
| verbose | Display progress | Optional | boolean |
True(Prints intermediate results) |
-
Create model of FiRE.
# model <- new(FiRE::FiRE, L, M, H, seed, verbose) model <- new(FiRE::FiRE, 100, 50, 1017881, 5489, 0)
| Parameter | Description | Required or Optional | Datatype | Default Value |
|---|---|---|---|---|
| L | Total number of estimators | Required | int |
- |
| M | Number of features to be randomly sampled for each estimator | Required | int |
- |
| H | Number of bins in hash table | Optional | int |
1017881 |
| seed | Seed for random number generator | Optional | int |
5489 |
| verbose | Controls verbosity of program at run time (0/1) | Optional | int |
0 (silent) |
-
Apply model to the above dataset.
model$fit(preprocessedData)Acceptable datatype is of
matrixclass and oftypedouble(Numeric matrix). -
Calculate FiRE score of every cell.
# Returns a numeric vector score <- model$score(preprocessedData) -
Select cells with higher values of FiRE score, that satisfy IQR-based thresholding criteria.
```R #Apply IQR-based criteria to identify rare cells for further downstream analysis. q3 <- quantile(score, 0.75) iqr <- IQR(score) th <- q3 + (1.5*iqr)
#Select indexes that satisfy IQR-based thresholding criteria. indIqr <- which(score >= th)
#Create a file with binary predictions predictions <- integer(dim(data)[1]) predictions[indIqr] <- 1 #Replace predictions for rare cells with ‘1’.
8. <h4>Access to model parameters.</h4>
Sampled dimensions can be accessed via
```R
# type : Integer matrix
# shape : L x M
model$d
Chosen thresholds can be accessed via
# type : Numeric matrix
# shape : L x M
model$ths
Weights can be accessed via
# type : Numeric matrix
# shape : 0 x 0
model$w
Hash tables can be accessed via
# type : List
# shape : L x H x <dynamic>
# <dynamic> : as per number of samples in a bin (H) for a given estimator (L).
model$b
Publication
Jindal, A., Gupta, P., Jayadeva and Sengupta, D., 2018. Discovery of rare cells from voluminous single cell expression data. Nature communications, 9(1), p.4719. DOI: https://doi.org/10.1038/s41467-018-07234-6
Copyright
This software package is distributed under GNU GPL v3.
This work is free to use for academic and research purposes. Please contact Dr. Debarka for commercial use of this work.
Patent
India Patent Office, Patent No: 582027 Finding Rare Samples in Given Dataset.